Chromatography
Approaches to troubleshooting an SPE method for the analysis of oligonucleotides (pt i)
Mar 13 2024
Author: Tony Edge on behalf of Avantor
Free to read
Articles are free to download. Unlock the article to be shown more content, graphs and images.
Solid phase extraction (SPE) is a powerful technique widely used in analytical chemistry to isolate and purify compounds from complex mixtures. It is often used for food and environmental samples, however, the most common application is in the field of bioanalysis, where it is ideally suited to removing the bulk of a complex biological matrix, whilst selectively extracting the compound(s) of interest. Whether you’re a seasoned professional or a beginner in the field, encountering issues during SPE is not uncommon. Troubleshooting problems in SPE is a systematic process, and understanding common pitfalls and solutions is essential to achieve reliable and accurate results. This article will look to identify some common issues that are associated with the use and practice of SPE, and look at how to address these challenges, through initial identification of the issue and then finally their alleviation to ensure the development of a robust methodology. As an example, the development of an SPE method for the analysis of an oligonucleotide mixture will be used to demonstrate some of the issues that an analyst can face. The original assay for the oligonucleotide mixture had poor reproducibility and also low recovery.
Understanding Solid Phase Extraction
SPE involves the selective retention of analytes on a solid sorbent, followed by elution for subsequent analysis [1] [2] [3]. A typical SPE cartridge will comprise of a sorbent, which will have a bed mass, and also a defined volume to contain the various solvents and samples that are used throughout the extraction process. The maximum loading capacity of the bed mass will typically be about 5%. The volume of the cartridge will typically relate to the sample volume being used and also how much sample will need to be passed through the sorbent bed to obtain suitable detection limits [4] [5]. Both sorbent mass and cartridge volume will be optimised as part of the method development process. The extraction process typically includes these steps:
Conditioning: Preparing the SPE cartridges or plates with a suitable solvent to activate and equilibrate the sorbent, since the sorbent will typically be in a dry state before it is used. This step is critical to ensure that the full surface area is available for the adsorption of the analyte. Many of the sample types are aqueous based and the majority of the commonly used SPE stationary phases are hydrophobic in nature and so having some organic solvent present will ensure that the sample can get access to the full surface area.
Re-equilibration: One of the disadvantages of applying the organic solvent is that it acts as a very strong eluting solvent and so it is necessary to remove the bulk of this organic solvent by the addition of a re-equilibration solvent, or a very low elution strength solvent. This will not displace all of the organic solvent but it will be at such a low concentration that it will not be strong enough to elute at the next stage (loading). The pH can also affect the ability of a solvent to elute the compound of interest as it will nominally change the charge state that the compound or the stationary phase exists in.
Loading: Introducing the sample to pass through the SPE media, allowing the analytes to bind to the sorbent selectively. Many critical parameters can affect the performance of this stage including;
Stationary phase selection. There is a range of stationary phases available and understanding how those phases interact with the analytes is essential for good recovery and will also determine how to optimise pH and solvent selection.
Many silica and polymeric-based stationary may have a range of secondary interactions that exist and having a full knowledge of these is critical to ensuring high recoveries are obtained.
The selection of an appropriate pH selection can be critical to optimising the analyte retention at the phase of the method [4], as many analytes have ionisable groups that will become charged depending on the pH, and this will affect the polarity of the molecule but also the possible interactions with the stationary phases.
The flow rate can also have a significant impact on the adsorption mechanism, and ideally slower loading flow should always be applied.
Washing: Removing as many interfering compounds as possible by washing the sorbent bed with solvents of varying polarity. This involves the optimisation of both pH and the nature of the solvent, typically it will be the organic component. The washing should remove as much of the less retentive matrix components as possible without removing the analyte. Performing multiple washes will optimise the removal of different types of components, that will be impacted by changes in the pH and organic solvent composition, for example.
Elution: Recovering the analytes of interest by using an elution solvent that disrupts their interaction with the sorbent. There are two considerations when optimising the elution step, relating to the retention of matrix components on the sorbent and also any subsequent steps, primarily a blow down step, which happens after the elution stage. Applying too strong an elution solvent may result in co-elution of an increased number of matrix components with the target analyte, resulting in increased matrix affects when performing the final chromatography. It is common when using SPE to blow down the elution solvent, and aqueous components can substantially increase the length of the blow down stage, typically by a factor of 4 – 5, which often means that many analysts will default to the use of nearly 100% organic elution solvent.
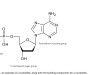
In any method development, the initial stages are to create an experimental framework that identifies a suitable approach to the extraction process and to highlight any potential issues that may arise as the method is being developed. An oligonucleotide is an oligomer based on a handful of nucleotides. The nucleotides comprise of three major groups which are; phosphate, a five membered sugar group and a nucleobase (either a purine or a pyrimidine), an example is given in Figure 1, along with the possible building components.
Figure 1. An example of a nucleotide, along with the building components for a nucleotide.
The original SPE method utilised a weak anion exchange phase (WAX) for extraction of the oligonucleotides, which was based on a generic small molecule anion exchange protocol . A weak anion exchange stationary phase has a positive charge associated with it and is typically obtained using an amine functionality, which has a pKa of approximately 9. This will vary depending on the manufacturer and also the exact nature of the phase and the substrate material. A weak anion exchange phase will have a positive charge below the pKa and it will be neutral above the pKa. With the oligonucleotide, it is the phosphate functionality that will be targeted. This functional moiety has a negative charge across the full pH range making it ideal for trapping on a weak anion exchange phase.
It should also be noted though that the nucleobase and sugar moieties potentially complicate the possible interactions that can exist between the analyte and the stationary phase, since there is a potential for some hydrophobic, hydrophilic and even cation exchange interactions, depending on the nature of the stationary phase and the sorbent material. Thus, a silica substrate has a weakly acidic functionality which could interact with the positively charged nucleobase [6].
The initial method used a standard protocol, given in Table 1. This protocol relies on the pH of the solvents being employed to alter the charged state of the sorbent material, so at a low pH the sorbent material is charged and at a high pH, greater than the pKa the material is in a neutral state.
Table 1. The original methodology is based on a generic method, using a 30 mg bed mass, with a 2 mL sample volume format.
Step Solvent
Condition 1 mL 0.1% formic acid in methanol
Re-equilibrate 1 mL 0.1% formic acid in water
Load 1 mL 30 µg/mL oligonucleotide in 0.1% formic acid in water
Elute 2 x 0.5 mL 5% ammonia in methanol
Figure 2 shows the initial chromatograms obtained when using this protocol with an aqueous solution of the oligonucleotide. It is very evident that the SPE protocol is not working and that there is something wrong with the understanding of the retention mechanism of the extraction sorbent.
Figure 2. The chromatography obtained using the original protocol compared to a standard of nominally the same concentration.
The generic methodology (Table 1) was originally developed for a small molecule acid, in this case salicylic acid and it was noted that the recovery for the compound was good and the standard protocol worked well. It is evident then that there is an issue with the understanding behind the retention mechanism of an oligonucleotide on a silica based WAX phase.
The initial hypothesis was that the zwitterionic oligonucleotide had multiple retention mechanisms with both acidic and basic groups interacting with the stationary phase sorbent and the substrate, which have opposite charges associated with them, and this resulted in poor recovery and poor elution. Figure 3 shows the recovery obtained using an aqueous solution of salicylic acid using the standard protocol on the WAX sorbent at the loading and elution stages. It is evident that the phase is working and that using the standard protocol the salicylic acid is retained and then eluted under the methodology prescribed in Table 1.
It is known that to perform the chromatography, the use of ion pairing reagents had to be implemented to get good peak shape and also consistent retention times for a silica based column (ref), thus the first set of troubleshooting experiments investigated the impacts of the addition of TEA (triethylamine) and varying amounts of organic has on the final elution stage. The SPE method conditions are presented in Table 2, whilst Figure 4 showing the results obtained. It can be seen that the highest recovery was obtained using 100% aqueous elution solvent with 50 mM TEA added. It is evident that the addition of organic solvent has an impact.
Table 2. The revised method using an ion-pairing reagent to aid elution from the WAX SPE media.
Step Solvent
Condition 2 x 1 mL methanol
Re-equilibrate 2 x 1 mL 50 mM ammonium acetate in water
Load 1 mL 30 µg/mL in 50 mM ammonium acetate in water
Elute 4 x 250 µL 50 mM TEA
Figure 4. The addition of TEA to the method substantially improved the recovery.
With the success of the use of TEA, the next set of experiments looked at the impact of varying the amount of TEA added to the elution solvent. It can be seen in Figure 5, that increasing the concentration of the TEA has a pronounced effect on the recovery, with nearly 100% recovery being achieved when eluting with 400 mM TEA compared to no oligonucleotides being present in the elution solvent, when there is no TEA added. This strongly suggests that the interaction of the substrate is having a big impact on the overall assay performance.
Figure 5. The effect of varying the concentration of TEA on the recovery an oligonucleotide.
Once the optimal concentration of TEA was determined, a further set of experiments was performed to investigate the volume of elution solvent that was required to obtain the maximum recovery. In this experiment, the elution volume was reduced to 500 µL and multiple elutions were performed to indicate how much volume was required. Figure 6 clearly shows that two volumes of 500 µL are required to remove the majority of the oligonucleotide, and confirmatory experiments demonstrate that increasing the number of elutions to 4 or 6 has no substantial impact on the recovery, since it has already achieved 100%.
Figure 6. Determine what volume is required to obtain the highest recovery, using 200 mM TEA in the elution solvent.
Table 3 gives the initial and final methods for the extraction protocol.
Table 3. Initial and final methods for the extraction protocol.
Initial method Final Method
Step Solvent Step Solvent
Condition 1 mL 0.1% formic acid in methanol Condition 2 x 1 mL methanol
Re-equilibrate 1 mL 0.1% formic acid in water Re-equilibrate 2 x 1 mL 50 mM ammonium acetate in water
Load 1 mL 30 µg/mL oligonucleotide in 0.1% formic acid in water Load 1 mL 30 µg/mL in 50 mM ammonium acetate in water
Elute 2 x 0.5 mL 5% ammonia in methanol Elute 4 x 250 µL 50 mM TEA
Conclusion
It is evident that careful consideration of the analyte properties and also the sorbent bed properties is critical to ensure that optimal recovery is established when utilising SPE. When the analyte molecule becomes larger, there is a tendency for a greater number of potential interactions to occur between the stationary phase and the analyte molecule as was seen when troubleshooting the SPE method for an oligonucleotide. In this scenario, the use of a methodology which was demonstrated to work for a simple organic acid is no longer applicable when targeting the more complex molecule. This resulted in poor recovery. Consideration of the structure identified that the multiple active moieties on the stationary phase and the analyte meant that an ion pairing approach to the extraction would be better as this negated some of the adverse interactions that resulted in poor recovery of the assay. More importantly it was shown that by having a systematic approach to troubleshooting and progressing through the method optimisation steps in a methodical manner, ensured that not only was higher analyte recovery achieved, but also a better understanding of the parameters that cause the instabilities. This approach could be applied to the troubleshooting of any analyte on any stationary phase and the number of experiments and the time taken to perform these experiments was not prohibitive, which suggests that this may be a better approach to not only troubleshooting an extraction process but also in terms of optimising or developing a methodology. Too often standard methodologies are employed without an understanding of the basic chemistry and separation process that drives the extraction process. An understanding of the chemical interactions not only aids method development but also ensures that the methods are inherently more robust.
References
1. C. F. Poole, Solid-phase extraction, Elsevier, 2020.
2. V. Samanidou, Solid Phase Extraction: State of the Art and Future Perspectives, Multidisciplinary Digital Publishing Institute, 2019.
3. M.-C. Henion, “Solid-phase extraction: method development, sorbents, and coupling with liquid chromatography,” urnal of Chromatography A, vol. 856, no. 1-2, pp. 3-54, 1999.
4. D. E. Raynie and D. W. Watson, “Understanding and Improving Solid-Phase Extraction,” LCGC North America, vol. 32, no. 12, pp. 908-915, 2014.
5. G. J. Maranata, N. O. Surya and A. N. Hasanah, “Optimising factors affecting solid phase extraction performances of molecular imprinted polymer as recent sample preparation technique,” Heliyon, vol. 7, no. 1, pp. 1-12, 2021.
6. J. Nawrocki, “The silanol group and its role in liquid chromatography,” Journal of Chromatography A, vol. 779, no. 1-2, pp. 29-71, 1997.
Free to read
Articles are free to download. Please login to read this article or create an account.
Digital Edition
Lab Asia 31.2 April 2024
April 2024
In This Edition Chromatography Articles - Approaches to troubleshooting an SPE method for the analysis of oligonucleotides (pt i) - High-precision liquid flow processes demand full fluidic c...
View all digital editions


.jpg)






Events
Apr 28 2024 Montreal, Quebec, Canada
May 05 2024 Seville, Spain
InformEx Zone at CPhl North America
May 07 2024 Pennsylvania, PA, USA
May 14 2024 Oklahoma City, OK, USA
May 15 2024 Birmingham, UK





