Laboratory products
Published over 6 years ago. See the latest and most current information on Laboratory products.
Despite advances in genomics, some aspects of cell behaviour can only be understood by exploring proteins. Cell signalling pathways, which commonly rely upon protein ‘receptors’ binding to signalling molecules to enable communication, allow a cell to interact with and respond to its environment appropriately, supporting healthy and normal growth, migration, division, and development and repair of tissue. These pathways can break down or become disrupted by various genetic or epigenetic alterations, leading to irregular cell function and the development of tumours and cancers.
To better understand the mechanisms behind cell health and disease, researchers aim to determine how cell signalling pathways are disrupted, and the points at which they are impeded along the sequential signalling pathway. Proteomics is a valuable tool here, as it can provide a level of information that other modalities cannot. By targeting specific proteins, proteomics directly measures the components of a cell rather than evaluating by inference based on RNA, DNA or other biomolecules. Quantitative proteomics advances these capabilities and identifies not only what is in a cell, but how much of it is present and how different proteins are interacting, providing a ‘systems level’ understanding of what is going on in a given cell or disease state.
While it generates invaluable insight, proteomics remains challenging due to the sheer complexity of the proteome. While the genome is made up of approximately 20,000 genes, the proteome comprises up to one million protein forms, some of which are present at low (and, therefore, hard-to-detect) abundances. The proteome is also highly dynamic and variable in nature; it responds to environmental influences and changes with age and individual characteristics, making sampling and interpretation difficult. Additionally, many laboratories have not yet fully embraced proteomics, considering it to be a somewhat specialised approach.
However, the drug discovery landscape is changing, with innovative analytical technology playing an increasingly vital role at this stage of the pipeline and removing traditional barriers to entry. Advanced proteomics methodologies offer more accessible, usable ways to characterise the complex molecular mechanisms involved in disease pathogenesis – without compromising on performance.
Quantitative proteomics aims to paint a reliable, accurate picture of the proteome - of its protein concentrations, post-translational modifications, protein-protein interactions, and more - in order to understand the molecular drivers of disease. As the proteome is the main functional entity within a cell, proteomic analysis of cells in various disease states can identify potential biomarkers for use in novel biotherapeutics, and enable researchers to better understand the mechanisms underpinning diseases, such as cancer (which, in turn, could unlock faster diagnosis and treatment for patients). While oncological research is a notable example of the value of proteomics, quantitative proteomics holds promise in many other areas of study, from the development of wider biotherapeutics and personalised medicines to metabolomics, anti-doping, food and beverage testing, and more.
Traditionally, clinical diagnosis relies upon a mix of pathology, microscopy and genomic stratification – but this is insufficient for optimal understanding and diagnosis of health and disease given that many drugs act on proteins. With quantitative proteomics, diagnostic or prognostic protein biomarkers can be derived and used to monitor a specific set of targets, and map signalling networks and pathways. Such pathways are contextual in nature; measuring a specific node may return a specific set of conditions, but this gives just a snapshot of a particular pathway. For instance, a particularly active protein may appear to be in a certain state, but it may in fact be manipulated by a node further along the pathway. In this way, reaching a conclusion based on just one aspect of a sample’s total suite of proteins may return false conclusions and, in turn, prompt incorrect clinical research decisions – or in a diagnostic setting, incorrect decisions regarding patient treatment. Additionally, it may be easy to measure the most visible proteins in a cell, but some signalling pathway components exist only in very low abundances. As with an iceberg, the ‘tip’ of higher-abundance proteins may be visible - but what if the important information lies beneath?
Despite its promise, quantitative proteomics requires advanced technology. A key issue relates to the aforementioned dynamism and variability of the proteome. This is far more pronounced than for the genome, and can enhance sampling bias. Sampling different parts of the same tumour results in differences, and methods of collection and storage become important considerations since, for example, post-resection freezing delays can impact the activation of signalling networks. In such cases, researchers are not seeing the real physiology of the disease, but the artefacts of collection and biobanking. Issues of sampling bias, or of changes occurring during collection and storage, do not arise for genomics samples as the DNA is stable. The comparative instability of the proteome, and associated sample variability, highlights the challenges inherent in oncology and disease proteomics.
Currently, quantitative proteomics comprises a combination of discovery-based and targeted methods. Discovery methods aim to comprehensively survey a sample and identify all the components present, while targeted approaches instead seek to monitor and quantify selected targets of interest.
Many quantitative proteomics methodologies are of the former type – they look more broadly at the whole system – and are based on antibodies. Predominant antibody-based protein measurement methodologies include Luminex, tissue microarrays and western blotting. These techniques seek to determine which antigens within a sample are binding with specific and selective antibodies.
However, antibody-based methods are limited by the quality, availability and selectivity of the desired antibody, and can typically explore a maximum of several hundred or so targets. Mass spectrometry (MS), meanwhile, can explore thousands of targets without needing to verify the quality of an antibody, raising confidence in results. More targeted methods of quantitative proteomics use MS – specifically, liquid chromatography MS with ‘selected reaction monitoring’ triple quadrupole workflows (LC-MS; SRM). An example of such a workflow is one in which peptides representing proteins of interest are used to generate an ‘assay’ for their detection and quantification. For this, selected peptides are isolated and fragmented and characteristic fragment ions for that peptide are then sequentially isolated and detected. By tracing the properties of these precursor-product ion pairs, the target peptide – and, by extension, the target protein – can be quantified.
Quick, straightforward and cost-effective SRM approaches are limited by mass resolution and selectivity, especially in samples with complex matrixes such a biological material. However, such limitations can be overcome using high-resolution, accurate-mass MS (HRAM MS) and parallel-reaction monitoring workflows (PRM), such as those implemented by Thermo Scientific Orbitrap-based mass spectrometers. PRM isolates a target precursor, fragments it and then detects all resulting product ions simultaneously, allowing quantification of peptide abundance and comparison of results across multiple sample sets.
This brings greater selectivity and sensitivity alongside lower limits of detection, but remains limited by speed of acquisition, throughput and sometimes representation of the amount of target substance present (a property defined as peak area). Peak area is limited for PRM due to its slower acquisition, which decreases the chances of it achieving complete sampling of a peptide’s elution profile.
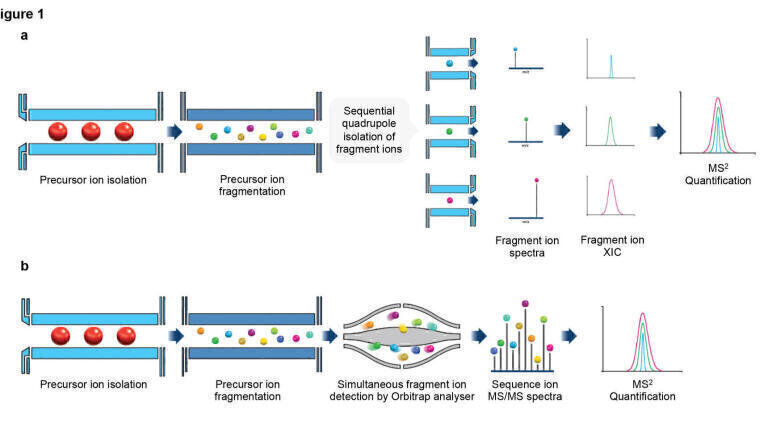
SRM and PRM are conventional targeted LC-MS approaches. (a) In SRM assays the mass spectrometer is programmed to monitor for the presence of one or more precursor ions, collisionally fragment these ions, isolate, and detect the resulting fragment ions in sequential steps. The integration of extracted ion chromatograms (XIC) from the diagnostic fragment ions allows quantitation of the target. (b) PRM shares similarities with SRM, however, fragment ions are isolated and detected in parallel using high-resolution, accurate mass detectors such as Orbitrap mass analysers. This enables post-acquisition determination of the optimal fragment ions for quantification as well as higher measurement selectivity.
For both SRM and PRM, a major constraint lies in the relationship between target multiplexing (the number of analytes that can be measured reliably) in the desired cycle time and the amount of time devoted to each analyte within that timeframe. Cycle time is constrained by the properties of the LC setup, and so there is always a tradeoff between the highest performance (in terms of selectivity and sensitivity) and number of targets per analysis. Increasing the amount of time devoted to a single analyte brings improved sensitivity, for instance, but allows fewer analytes to be quantified. Conversely, large numbers of targets cannot be studied for as long, compromising data quality. Overall, LC-MS approaches either bring high-scale coverage with sub-optimal quantitation, or low-scale coverage with high quantitation, but not both.
Inter-dependencies between experiment scale, sensitivity, and selectivity. Chromatographic elution properties of the analyte and the desired sample rate (left) will ultimately determine the amount of time the mass spectrometer can spend taking measurements (centre). Within this fixed time, the instrument can be used to collect fewer measurements with high selectivity and sensitivity or alternatively, take more measurements but a reduced sensitivity (right).
The limitations of common LC-MS and antibody-based quantitative proteomics methodologies are far from abstract: they have real-world impacts on patients. They result in new therapeutic options either being delayed or unavailable, due to a limited understanding of how tumours and diseased cells sign al, and how best to target them via treatment.
To overcome these hurdles, a new approach – internal standard triggered PRM (IS-PRM) – was developed that can dynamically guide targeted analysis in real time. This allows large numbers of targets to be reliably measured at high sensitivity and selectivity, protecting data quality and improving the efficiency of the analytical process. However, early iterations of IS-PRM require an advanced level of technical knowledge, with the user needing to utilise complicated programming interfaces and informatics tools to develop assays and prepare methods that achieve only a partial implementation. This has limited the uptake of IS-PRM by the proteomics community.
More recent developments remove this barrier. The Thermo Scientific SureQuant IS Targeted Quantitation method, for instance, is an evolution of the original IS-PRM approach. It presents the methodology in a usable, easily implementable way as a turnkey ‘off the shelf’ solution for any laboratory, including for use in biotherapeutics, plasma analysis, host-cell protein analysis, and to replace traditional biochemical analytical approaches such as ELISA.
Targeted proteomics workflows have traditionally been challenging, especially for people new to the technology, as the amount of time and effort needed to develop, standardise and validate a targeted assay can be considerable. IS-PRM technologies, such as the SureQuant method, simplify this by bringing an intelligent approach to proteomics, which in turn maximises chances of successful detection and quantitation.
SureQuant intelligent detection of targets maximises instrument efficiency and productivity. The IS and endogenous detection of a representative peptide, LCDSGELVAIK, is shown from PRM and SureQuant acquisition. In the PRM experiment, many uninformative MS2 scans are captured for the IS and endogenous target (grey region) during the 2.5 min monitoring window, and a smaller proportion of MS2 scans are captured during the actual target elution time (white region). The dynamic nature of SureQuant acquisition minimizes unproductive scans allowing shorter duty cycles and higher productivity. Experiment details: 50 fmol IS spiked into 250 ng HeLa cell line digest. PRM MS2 settings: 2.5 min RT window, 15000 resolution, 20 ms IT. SureQuant MS2 settings: Watch mode 7500 resolution, 10 ms IT; Quant mode 60000 resolution, 116 ms IT.
Refined IS-PRM approaches such as SureQuant bring superior acquisition efficiencies of 80-90% (cf. 10-15% via other conventional targeted approaches), as acquisition parameters can be adjusted on-the-fly to maximise sensitivity and selectivity at the time-point when target is eluting. This productivity brings enhanced data quality and allows more targets to be quantified in the same amount of analysis time to increase target scale and throughput. Importantly, it enhances the chance of successful detection, as the use of internal standards intelligently guides measurement of the target of interest at precisely the right time.
The chances of missing a target measurement are, therefore, greatly minimised, resulting in more consistent, reliable and robust measurement and quantitation. Obtaining a more reliable and detailed dataset allows researchers to make fully informed decisions, and can help to meet compliance needs in highly regulated environments by enabling more precise quantification of the substances within a drug product.
SureQuant acquisition robustness overcomes chromatographic fluctuations. Targeted analysis of AKT-mTOR pathway proteins was performed by PRM and SureQuant acquisition using a standard gradient and an offset gradient which introduced a 5 minute artificial time delay to simulate LC retention time variations that can commonly occur (landmark peptides A-D are indicated for comparison). As an example, the observed heavy peptide LFDAPEAPLPSR (m/z 611.8526 ++) elution is shown at the original and offset retention times (bottom left). Notably, while PRM acquisition failed to capture the signal of the peptides with delayed elution, SureQuant acquisition maintained reliable measurement under these conditions.
Low-level cell signalling pathway modifications, such as tyrosine phosphorylation (pTyr) on proteins, play a key role in cell signalling, and have been found to be commonly dysregulated in cancer cells. Profiling the tumour pTyr proteome may, therefore, reveal insights central to the development of therapeutic oncology treatments. However, traditional MS-based methods do not offer adequate sensitivity, reproducibility and depth of coverage given that pTyr proteins account for less than 1% of cellular phosphorylation.
To overcome these challenges and enable research into phosphoproteomics, Professor Forest White’s research group at the Massachusetts Institute of Technology (MIT), implemented SureQuant methodology, achieving reproducible and sensitive targeted quantitation as a result. White used this technique to leverage isotopically-labelled trigger peptides and reliably quantify several hundred commonly dysregulated pTyr targets in human colorectal tumour samples.
“SureQuant is uniquely positioned for phosphotyrosine tumour studies,” said White, Professor of Biological Engineering at MIT. “Targeted approaches typically allow observation of dozens of targets at one time, but the SureQuant setups available instead allow thousands - and the number of pTyr events within the cell likely number in the same range, with at least 2,000 sites. We aim to expand our SureQuant work to develop a larger-scale model that can cover all functionally important pTyr sites, so we can monitor the phosphotyrosine signalling network.”
Quantitative proteomics methodologies are essential tools in the drug discovery toolkit. Discovery-based and targeted methods of quantitative proteomics are perceived as having opposing aims, with one type positioned to take a broader view and one a more detailed view. However, novel and refined methods of IS-PRM are capable of achieving large-scale target profiling with superior quantitative performance, filling a critical gap in protein quantification workflows. As well as enabling a deeper understanding of the molecular mechanisms behind cancers, such technology has great potential beyond oncological proteomics. The SureQuant method promises to play a crucial role in advancing our understanding of a wide range of complex diseases and signalling pathways, unlocking a wealth of knowledge about cell behaviour and health - and signposting potential routes to timely, effective treatments.
Lab Asia 33.4 - August 2026













-(1).jpg)


.jpg)





.jpg)






