Mass spectrometry & spectroscopy
Published over 4 years ago. See the latest and most current information on Mass spectrometry & spectroscopy.
‘How much drug is actually inside my drug?’ is an important question drug manufacturers ask on a routine basis. Transmission Raman Spectroscopy (TRS) is an excellent technique to answer this question. TRS can be used to analyse whole intact tablets and capsules in seconds; to generate a bulk quantitative result without sample preparation. This tutorial article is a summary of the method development process.
Transmission Raman Spectroscopy (TRS) is a regulatory approved method for Content Uniformity (CU) testing. Content Uniformity (CU) is a mandatory batch release test for oral solid dose (OSD) forms such as tablets and capsules. It is defined by USP <905> Uniformity of Dosage Units [1].
TRS is a fast, bulk, non-destructive technology which presents an efficient and cost effective complimentary analytical workflow in the Pharmaceutical QC laboratory. This technique is not only effective in terms of speed (less than 1 minute per sample) but also efficient in terms of entirely negating sample preparation solvents and consumables.
Often the route of implementation of spectroscopic methods is not well understood, despite being well referenced and supported by industry guidance [2,3,4,5]. This article sets out the main steps of implementing a spectroscopic technique as a batch release test.
1. Feasibility
2. Calibration
3. Validation
4. Reference measurements & model building
5. Implementation
6. Model maintenance
Although the prominent application of TRS is for CU testing, further quantitative application areas in real time release testing, in-process check, formulation development, crystallinity and polymorph analysis are also possible. The quantitative method development process is analogous between applications.
Before a large analytical method development workflow is undertaken, it is important to understand if the application will work. Feasibility studies include scanning many different products that may be suitable and looking for a formulation and product form that suits the technology. In general, a formulation with >1% w/w API (active pharmaceutical ingredient) content as a tablet or capsule is suitable. Higher concentrations between 10-40% w/w will be easier as the API contributions in the spectra will be stronger. Other forms of products such as powders, creams and solutions are also viable applications.
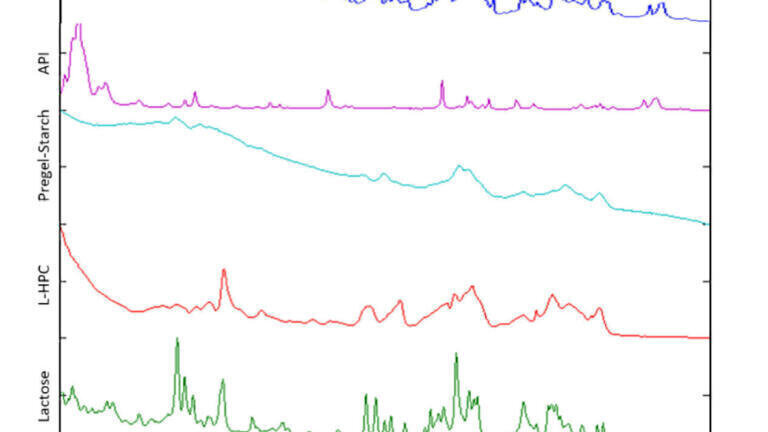
Feasibility testing should start with analysis of the final product and pure components to ascertain if good quality spectra can be obtained. It is essential that the API and main excipients are visible in the final product spectra. Clearly defined Raman spectra with low levels of fluorescence are ideal properties for a good candidate. An example is shown in Figure 1, where the API peaks ~700, 1100 and 1600 cm-1 are clearly visible, along with the excipient (lactose) peaks around 300-500 cm-1.
At this stage a small calibration sample set may be built to further investigate the viability. This may be a spiking study of API into a placebo blend; but this is not a complete calibration.
At this stage looking at historical batches of a product to obtain a proper feel for the batch-to-batch spectral variation (if any) is also recommended.
The next stage is calibration, which involves making samples of different concentration and building a chemometric model. Commonly used chemometric models for bulk quantitation of this nature involve Principal Component Analysis (PCA), Particle Least Squares (PLS), Partial Least Squares Discriminant Analysis (PLS-DA).
Calibration samples should be made following a design of experiment (DoE) that encapsules the expected process and product variation in factors which are deemed high risk with respect to spectral variability. For example, one may expect API and the main excipients to change in the DoE; but unlikely to expect minor excipients/lubricant or particle size distribution to vary. As part of the calibration design, it’s recommended that a risk-based approach is carried out to ascertain which factors affect the specifics of the product being analysed.
A typical calibration will be between 7 to 25 samples compressed into n number of tablets where at least 3 tablets, ideally more, are analysed per sample.
Acquisition parameters will be optimised to achieve satisfactory Raman signal in as shortest time as possible, by altering laser power and scan time.
Multiple measurements per sample can also be obtained. Measurement times per sample are generally < 1 min. Typically transmission Raman spectra of small white tablets require a ~20 second measurement.
Calibration spectra are then used in the model building (See section 4.)
Validation samples are essential for testing the calibration. Validation samples are independent from the calibration. Validation samples can be a mix of laboratory made and production material. Validation samples should adequately test the calibration design for it’s given application and be representative of the process that is intending to be measured.
These samples will be analysed as a confirmatory test to ensure that the model works.
A typical validation sample set would be 10 to 20 separate batches and at least 10 samples per batch.
Once all the calibration and validation Raman spectra have been acquired, reference measurements of the samples are then acquired using the primary technique. The primary analytical method is almost always HPLC (High Performance Liquid Chromatography), but it could be another technique e.g., UV. The reference values are used as the ‘true’ values of the sample. This removes any ambiguity over sample preparation and tablet to tablet variability.
A chemometric model is used to translate a Raman spectrum into a ‘prediction’ or result. In this workflow the result is a prediction of the reference technique, which as mentioned earlier, is typically the HPLC’s result of that sample.
Industry guidance for chemometrics is thoroughly described in USP <1039> Chemometrics [6].
For quantitative TRS applications Partial Least squares (PLS) is the most common quantitative multivariate model used. As part of the implementation spectral preprocessing is performed to exemplify spectral variation due to the API and minimise spectral contributions from irrelevant parameters. Pre-processing is often a baselining followed by a normalisation and then mean centring. Spectral regions are also refined. The model building process is an iterative process and varies on an application basis.
As part of the model building process, the Raman prediction of the validation samples are compared to the HPLC reference values and an uncertainty is ascertained, as well as suitability of the spectroscopic method. The acceptance criteria of a given model can be application specific depending on the requirements and if often multi-faceted. In general, a 2% - 5% error of the Raman prediction compared to HPLC is often used.
An example of incorporating the ‘true’ values into the chemometric model is shown in Figure 2. The figure on the left is the gravimetric model and assumed the values of each of the individual datapoints (individual tablet) are the same as the bulk of the material it was prepared from. The model on the right is the Raman PLS model which incorporates HPLC values, where the HPLC values give a ‘true’ value for each tablet. In this scenario incorporating the true HPLC values into the model improves its performance.
Once confidence in the analytical method is achieved through rigorous feasibility testing, method development, verification and validation, the technique in question can be implemented. The regulatory requirements will differ depending on application e.g., an in-process check will differ from a CU batch release test. Methods can be used and approved via internal pharmaceutical quality systems (PQS) or by external regulatory authorities as required.
For any analytical method it is necessary to check it is working at set intervals; the same is true for multivariate spectroscopic techniques. Methods should be verified routinely, at time intervals deemed fit following a risk-based approach and checked according to the primary method. Models can be adapted and changed as the process changes and the change management can be via the PQS or external authorities.
1. USP Chapter <905> Content Uniformity Dosage Units
2. D. Andrews, K. Geentjens, B. Igne, et. al. J. Pharm. Innov. 13, 121–132 (2018).
3. J. Villaumié, D. Andrews, K. Geentjens, et. al. J. Pharm. Innov. 14, 245 – 258 (2019).
4. US Food and Drug Administration. Guidance Document; Development and Submission of Near Infrared Analytical Procedures Guidance for Industry. (FDA, Rockville, MD, 2021) FDA-2015-D-0868.
5. Römer, M. 4.5 Transmission Raman Spectroscopy - Implementation in Pharmaceutical Quality Control.
In Solid State Development and Processing of Pharmaceutical Molecules, M. Gruss (Ed.). (2021).
6. USP Chapter <1039> Chemometric.
ILM 51.5 July 2026



.jpg)
.jpg)









-(1).jpg)

.jpg)