Chromatography
Published over 4 years ago. See the latest and most current information on Chromatography.
Migration of methods between laboratories or from one analytical system to another when replacing technologies such as gas (GC) or liquid chromatographic (LC) system, either as a result of updating analytical equipment or changing from one supplier to another, could be a time consuming and difficult task. The instruments used in analytical laboratories are diverse and can belong to various brands. Often the same analytical method is used on instruments that are manufactured by different vendors with the expectation that the performance is equivalent.
As part of the method transfer and validation, federal and governmental agencies, such as the United States Food and Drug Administration (US-FDA), and the European Medicines Agency (EMA) released specific guidelines [1,2]. Moreover, the USP Chapter 621 of the current United States Pharmacopeia has suitability procedures to test analytical methods and demonstrate equivalency when transferring them from one system to another [3]. This is also applicable for GC methods where strict chromatographic separation criteria are defined.
In this paper, two examples of how the Thermo Scientific™ TRACE™ 1300 and 1600 Series Gas Chromatograph systems perform with typical, well-known GC methods for pharmaceutical and food industry are detailed, demonstrating the compatibility with common consumables such as liners and capillary columns, simplifying the method portability assuring equivalency of the analytical performance. The instrument conditions are not included herein. Please refer to the full paper (WP-74062) for information on instrument conditions.
Introduction
Solvents are widely used in the synthesis of pharmaceutical products, substances and excipients. To ensure patients’ safety, the International Conference on Harmonization (ICH) [4] and the United States Pharmacopeia (USP)[5] have published some guidelines to set the acceptable limits and to support the assessment of the residual solvents used during the production and purification processes. Residual solvents (RS) have low boiling points and thermal stability therefore they can be determined using headspace-gas chromatography (HS-GC) coupled to flame ionisation detection.
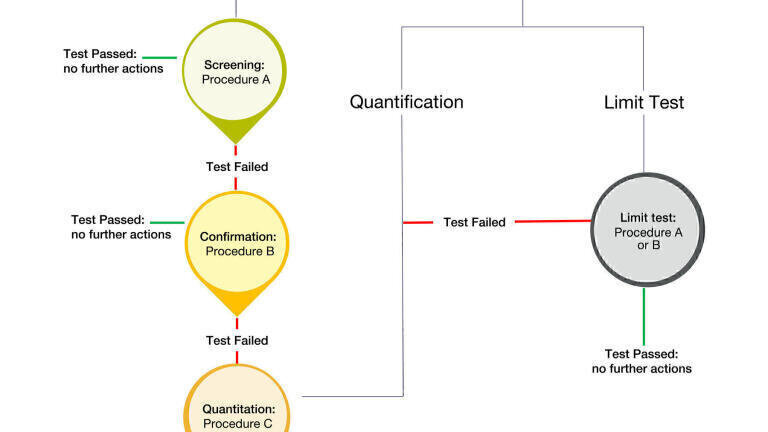
The workflow for residual solvent assessment is reported using a simplified schematic in Figure 1. When the residual solvents that are likely to be present are known, they can be determined using a limit test, such as Procedure A or Procedure B, or by a quantitative test, such as Procedure C. When the residual solvents are not known, then a screening test using Procedure A must be used. If the article does not meet the acceptance criteria of Procedure A, then Procedure B must be used to demonstrate compliance. If the article does not meet the criteria using Procedure A and Procedure B, then Procedure C must be used to quantify the residual solvents present in the article.
Procedure A - Screening of unknown residual solvents
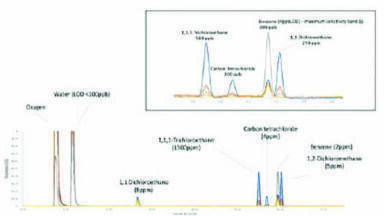
Stock, standard and test solutions were prepared according to the USP <467> method. An over-the-counter acetylsalicylic acid (dispersive aspirin, 75 mg) was purchased locally and analysed according to the USP <467> workflow in Figure 1.
System suitability criteria for sensitivity (peak-to-peak (PtP) signal-to-noise ratio (S/N)) and chromatographic resolution (Rs) were met with:
• S/N > 5:1 for 1,1,1-trichloroethane in Class 1 standard solution
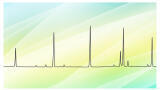
• S/N >3:1 for all peaks in Class 1 system suitability solution (Figure 2)
• Rs between acetonitrile/dichloromethane >1 in Class 2A standard solution (Figure 2).
The innovative system design with direct connection between the gas chromatograph and the autosampler combined with the high inertness and the precise temperature and flow controls of the TRACE 1310 Gas Chromatograph allowed for an efficient chromatographic process ensuring Gaussian peak shapes with average asymmetry factor (As) of 1.2. Peak responses obtained for the un-spiked sample were lower than the corresponding peaks in Class 1 and Class 2 standard injections. According to the regulation, the test solution met the requirements for residual solvent content with no other actions required.
A streamlined method transfer from a different HS-GC system using the Valve-and-Loop headspace technology is ensured by the consistency of the method parameters. The name to report the method parameters may differ within different brands, especially for the headspace autosampler. The equivalency of the parameters is clearly explained in a previous published white paper [6].
The results presented in this work demonstrate that the TRACE 1310 GC-FID fulfils the USP <467> requirements, meeting the suitability criteria for chromatographic separation as required for regulated c-GMP pharma laboratories. The equivalency of the method parameters assures a safe portability of the method from different HS-GC brands using the Valve-and-Loop headspace technology.
Separation of 37 fatty acid methyl esters according to AOAC method 996.06 by GC-FID
Introduction
Food fat mainly consists of triglycerides and assessing the fat (trans and saturated) composition of food products as part of the nutritional information is a fundamental test for the food industry. The AOAC method 996.06 describes the determination of total, saturated and unsaturated fat in foods using capillary GC-FID by a multiple steps procedure: hydrolytic extraction followed by the derivatisation (methylation) of fatty acids to produce fatty acids methyl esters (FAMEs) which are the derivatives suitable for GC analysis [7].
Experimental
A TRACE 1610 Gas Chromatograph configured with an Instant Connect split/splitless SSL Injector and an Instant Connect Flame Ionisation Detector (FID) was coupled with an AI/AS 1610 Autosampler and used to assess the chromatographic separation performance according to AOAC method 996.06.
A standard solution was prepared by diluting Restek Food Industry FAMEs mix (30 mg/mL in dichloromethane) (P/N 35077) to 1000 μg/mL in dichloromethane/hexane.
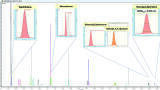
Chromatographic resolution (Rs) is fundamental for FAMEs separation, identification and quantitation and specific resolution requirements for critical peaks pair are included in AOAC method 996.06: (Rs) ≥ 1.0 for FAMEs pair of adjacent peaks (C18:3 - C20:1 and C22:1 – C23:3 – C20:4).
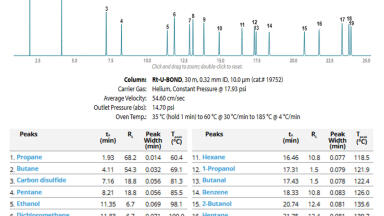
The chromatographic profile of 37 FAMEs separation obtained with TRACE 1610 Gas Chromatograph (equipped with Restek Rt-2560 column) is shown in Figure 2; critical pair peaks are highlighted, and the achieved resolution meets and exceeds the requirements. Peak identification and retention times are reported in Table 1.
The TRACE 1610 Gas Chromatograph equipped with Restek RT-2560 100 m, 0.25 mm, 0.2 μm capillary column is suitable for FAMEs separation in food samples according to AOAC method 996.06, meeting or exceeding resolution requirements and providing reliable peaks integration and quantification.
The examples considered in this white paper demonstrate that the TRACE Series Gas Chromatograph systems allows for equivalent chromatographic performance ensuring that suitability requirements of specific regulatory methods are met. The application of method parameters within a standard working range for the gas chromatographic system, along with the use of standard consumables, allow for a smooth transfer maintaining the required analytical performance.
References
1. EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2** Committee for Medicinal Products for Human Use (CHMP).
2. FOOD AND DRUG ADMINISTRATION OFFICE OF REGULATORY AFFAIRS ORA Laboratory Manual Volume II, ORA-LAB.5.4.5 Methods, Method Verification and Validation Revision #: 02 Revision June 2020.
3. USP Chapter 621 of the current United States Pharmacopeia.
4. Impurities: Guideline for Residual Solvents Q3C(R6), ICH Harmonised Guidelines, International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human use, 2016.
5. General Chapter USP <467> Organic Volatile impurities, Chemical Tests, United States Pharmacopeia, 2012 and Interim Revision Announcement Official November 1, 2019; Official December 1, 2020 <467> Residual Solvents.
6. Thermo Scientific White Paper 10705- Investigation of key parameters for a smooth method transfer to the new Thermo Scientific TriPlus 500 Headspace Autosampler
7. AOAC 996.06-1996 (2010) - Fat (Total, Saturated, and Unsaturated)
Please refer to WP-74062 for the full paper which will include:
- Determination of gasoline range organics (GRO) in water by static headspace gas chromatography
- Separation of US EPA 16 priority polycylic aromatic hydrocarbons by GC-FID
For the full paper (WP-74062), please contact [email protected]
Lab Asia 33.4 - August 2026




.jpg)









-(1).jpg)