Mass spectrometry & spectroscopy
Published over 7 years ago. See the latest and most current information on Mass spectrometry & spectroscopy.
NMR spectra provide various types of information, usually focused on the identification of the compounds contained in a sample. However, it is well-known that quantitative information can also be obtained. There are in fact 2 types of quantitative analysis in NMR:
• Relative quantitation: the measurement of the ratio of a target component contained within the sample being measured
• Absolute quantitation: measurement of the actual amount of the target component contained within the sample being measured.
Absolute quantitation has been attracting attention recently because it has some unique characteristics and advantages over other analytical methods. Chromatography detects the characteristics of the molecule itself, like the absorbance, refractive index, and fluorescence. This means that for quantitative analysis, we need a standard substance that is identical to the component that is being quantified in order to have a reference or yardstick for measuring the target molecule.
NMR detects the nuclei that form molecules. Consider hydrogen atoms in a molecule. Nearly all organic compounds contain hydrogen. As long as there is a proton in the molecule we are trying to quantify, we don’t need our reference yardstick to be a standard substance that is identical to the target component; it may be any suitable compound. This is a significant advantage and is one of the reasons that qNMR has been attracting attention.
NMR can be used for almost any organic compound that can be made into a solution. It can be applied to agrochemicals, pharmaceuticals, food additives, standard substances, and other classes of chemical compounds. Substances difficult to analyse quantitatively using chromatography where there is no standard substance available, such as new compounds, or unknown materials, can be quantitatively analysed using qNMR. It’s possible to use one reference substance for the quantitative analysis of many measurement targets.
There is no need to create calibration curves for qNMR and no conditioning is required for performing a measurement. For a low molecular weight compound, several milligrams are required to make a measurement, but each measurement can be completed in about 10-15 mins. If an appropriate protocol is followed, qNMR can be used to perform SI traceable purity assessments, therefore the reliability of the results can be assured.
Measurement conditions for routine proton NMR are not suitable for quantitative analysis.
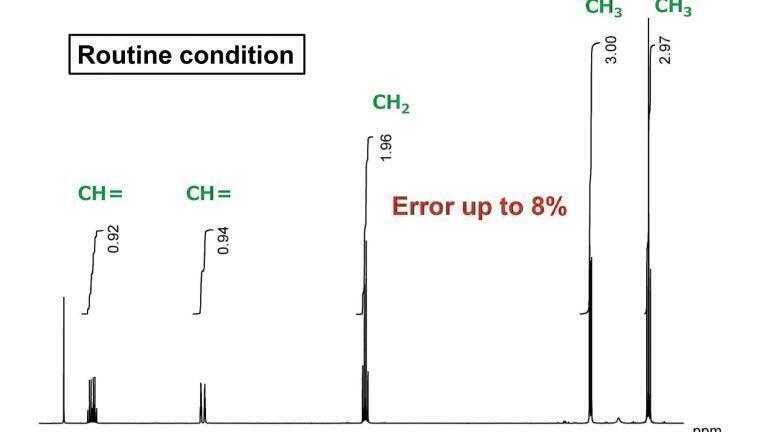
Figure 1 shows an NMR spectrum of ethyl crotonate acquired with ordinary, routine conditions, meaning the default measurement conditions on Jeol NMR instruments. They allow us to collect proton spectra quickly as required at many NMR laboratories. Figure 2 was acquired using measurement conditions optimised for quantification.
If we compare the peak areas, in Figure 1, from the left, the values are 0.92, 0.93, 1.96, 3.0 and 2.97. For a structural analysis, this can be interpreted as 1 to 1 to 2 to 3 to 3, but you can see that there is an error in the integral values of as much as 8%. This is unacceptable in high-precision qNMR analysis.
In comparison, in Figure 2, you can see that the area values and the proton counts match. The error is around 1%. This demonstrates the importance of using quantitative conditions when performing measurements for quantitative analysis.
Figure 3 shows a comparison of the typical parameters for routine conditions and for quantitative conditions. The routine conditions are the default settings of the Jeol instrument for proton measurement. The quantitative conditions are based on the conditions specified in the Japanese Pharmacopoeia. The parameter differences can be very broad, but there are 6 specific parameters that should be considered.
Typical parameters Routine Quantitative
Pulse repetition time ~7 sec >T1 x 7
Pulse flip angle 45° 90°
Scans 8 S/N>100
Digital resolution 0.5 Hz <0.25 Hz
Sample spinning On Off
13C decoupling Off On
Figure 3: Typical parameters for routine and quantitative measurement conditions
We will look in more detail now at the pulse repetition time and the number of scans which are two of the most important parameters.
As shown by Figure 4, the pulse repetition time is the length of time from the irradiation of one pulse until the irradiation of the next pulse. For quantitative conditions, this should be at least 7 times longer than T1 (longitudinal relaxation time).
Figure 4: Pulse repetition time overview
The magnetisation behaviour on the sample side corresponds to the pulse sequence of the instrument when the magnetisation is perturbed by the application of a pulse. If we wait long enough to allow the magnetisation to recover completely before applying the next pulse, the quantitativeness of the signals can be ensured. Therefore the parameter settings must ensure sufficient delay between pulses to ensure quantitativeness while making the measurements.
The index for setting this time is T1 – time constant that is a characteristic of the signal. The relaxation time can be determined by making an inversion recovery measurement. In order to determine how much time is required for the pulse repetition time, Figure 5 shows the theoretical relationship between the signal strength and the ratio between the repetition time, and the relaxation time. The vertical axis indicates normalised signal strength, and the horizontal axis is the ratio of the repetition time to the longitudinal relaxation time.
Figure 5: Pulse repetition time analysis
We can see the point at which the signal intensity turns to 100% expressed as a multiple of T1. According to this, setting the pulse repetition time to 7 x T1 or longer will mean the signal is nearly completely returned to the original state. This is why this is defined as the condition for quantification. In ordinary conditions, the main focus is on the signal to noise ratio in order to confirm the signal as fully as possible, meaning that the acquisition conditions emphasise the integration efficiency, so the goal is different.
The pulse repetition time is the most important parameter because it is the setting that theoretically improves the quantitativeness. The other parameters are mainly for minimising the integration error as we obtain the peak areas. There are no mandatory settings for the other parameters and there should be no problems if these are varied according to the situation.
For quantitative conditions, the signal to noise ratio should be 100 or more. Figure 6 shows the theoretical relationship between S/N ratio and the accuracy. According to this, if the S/N is 100 or more, the integration error can be kept to an accuracy within 1%.
In other words, to obtain an integration with a slightly better accuracy, the S/N ratio must be higher. If this cannot be obtained because of the sample amount, it is important to understand that this will be a factor contributing to the quantification error. Thus, the setting for the number of scans is not a specific number. It is a setting to obtain a target accuracy for the available S/N ratio.
Figure 6: Influence of signal intensity (S/N) to repetitive accuracy (SD) of integration
The analysis stage includes: data processing, calculation of purity and evaluation.
Appropriate data processing and signal selection, unlike measurement conditions settings, don’t provide a theoretical improvement. In principle, the integration range must be set to between 64 and 128 times the full width at half maximum of the signal in order to obtain the true integrated value of the signal, but this means setting an extremely wide range so this is unrealistic in most cases.
For signal selection, as NMR is not a separation analysis, it is necessary to consider whether there is any overlap of signals such as from impurities. There are various approaches for checking this. First, the integrals of the signals for all analytes must be obtained, each signal checked, and a final judgment made by looking at the variance between signals.
It is recommended to establish rules and to clarify the processing and signal selection used to obtain the numerical values. If the original spectrum is good, the errors in processing can be minimised. It is not a procedural point for processing, but making efforts to obtain good spectra during the measurement is important for successful qNMR.
qNMR analysis is becoming more and more attractive in a range of sectors thanks to its versatility. To optimise the measurement conditions, there are parameters that are universal for any case, but there are also parameters that must be considered for each sample. If challenges occur during analysis, it may be required to go back to the basics, so it is a good idea to establish a simple protocol before embarking on routine qNMR analysis.
Lab Asia 33.4 - August 2026




.jpg)









-(1).jpg)

.jpg)