Mass spectrometry & spectroscopy
Published over 8 years ago. See the latest and most current information on Mass spectrometry & spectroscopy.
Recent regulations on heavy metal testing have required the pharmaceutical industry to either monitor a suite of elemental impurities in drug products, or to implement a risk assessment strategy to show that their materials are free of these impurities. The PDE (Permitted Daily Exposure) of these 24 elemental impurity levels are defined in the new USP Chapter <232> [1]), and ICH Q3D, Step 4 guidelines [2]. Additionally, USP Chapter <233> suggests the use of plasma spectrochemistry to measure these elements in various drug delivery categories including oral, parenteral and inhalation. This article, adapted from one of the chapters in Robert Thomas’s new book, Measuring Elemental Impurities in Pharmaceuticals: A Practical Guide [3] offers guidance on which plasma spectrochemistry technique (ICP-OES or ICP-MS) is the optimum one to use for orally-delivered drugs, based on the J-values described in the validation protocols outlined in USP Chapter <233>.
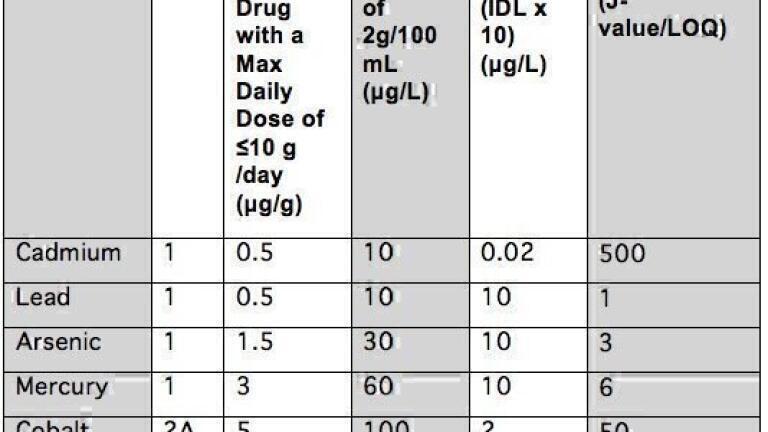
To get a better understanding of the suitability of the optimum plasma spectrochemistry technique being used and whether its detection capability is appropriate for pharmaceutical materials, it’s important to know the PDE limit for each target element. And in particular, what the USP calls the J-value, as described in Chapter <233>, which is defined as the PDE concentration of the element of interest, appropriately diluted to the working range of the instrument, after the sample preparation procedure is completed. So let’s take Pb as an example. The PDE limit for Pb in an oral medication defined in Chapter <232> is 5µg/day.
Based on a suggested dosage of 10 g of the drug product/day, that’s equivalent to 0.5 µg/g Pb. If 1.0 g of sample is digested/dissolved and made up to 500 mL, that’s a 500-fold dilution, which is equivalent to 1.0 µg/L. So the J value for Pb in this example is equal to1.0 µg/L.
The method then suggests using a calibration made up of 2 standards: Standard 1= 1.5J, Standard 2= 0.5 J. So for Pb, that’s equivalent to 1.5 µg/L for Standard 1and 0.5 µg/L for Standard 2.
The suitability of a technique is then determined by measuring the calibration drift and comparing results for Standard 1 before and after the analysis of all the sample solutions under test. This calibration drift should be < 20% for each target element. However, once the suitability of the technique has been determined in this way, further validation protocols described in detail in
It should also be pointed out that no specific instrumental parameters are suggested in Chapter <233>, but only to analyse according to the manufacturer’s suggested conditions and to calculate and report results based on the original sample size. However, it does say that appropriate measures must be taken to correct for interferences, such as matrix-induced wavelength overlaps in ICP-OES and argon-, matrix- and solvent-based polyatomic interferences with ICP-MS.
Let’s examine this by taking an example of measuring a suite of elemental impurities in an oral drug according to Chapter <232>, calculating the J-values for each elemental impurity and then comparing them with the limits of quantitation (LOQ) for ICP-OES, ICP-MS technique to give us an assessment of their suitability. For this analytical scenario, we’ll take the LOQ for the technique as 10x the IDL. These LOQs were calculated by taking the average of published IDLs from three instrument vendors’ application material and multiplying them by 10 to get an approximation of LOQ. In practice, a method limit of quantitation is typically determined by processing the matrix blank through the entire sample preparation procedure and taking 10 replicate measurements. The method LOQ, sometimes referred to as the method detection limit (MDL), is then calculated as 3-7 x standard deviations of these ten measurements, depending on the % confidence level required
To make this comparison valid, the sample weight was adjusted for each technique, based on the detection limit and analytical working range. So for ICP-OES we used a sample dilution of 2g/100 mL, whereas for ICP-MS we used 0.2 g/100 mL. ICP-OES could definitely use larger sample weights, but for high throughput routine analysis, we are probably at the optimum dilution for ICP-MS. (Note: For this assessment, it was felt that axial-ICP-OES was the better choice over the radial configuration, because of its superior detection capability).
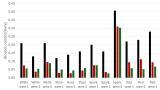
Tables 1 and 2 show the comparison of axial-ICP-OES and ICP-MS respectively for 7 elemental impurities defined as Class 1 and 2A elements in an oral drug with a maximum dosage of 10 g/day, according to Chapter <232>. The important data to consider is in the final column, labelled ‘Factor Improvement’, which is the J-value, divided by the LOQ. Generally speaking, the higher this number, the more suitable is the technique.
It’s important to note that the ‘Class’ column in these tables indicates the level of toxicity of the elements, which have been determined based on chronic exposure data and likelihood of occurrence in the drug product. The 24 elements in Chapter <232> and ICH Q3D are categorised into four classes - 1, 2A, 2B and 3. It is generally recognised that Class 1 and 2A elements are the most important to monitor, so this comparison will focus on these two groups of elements. Also it should be noted that the arsenic and mercury PDEs are based on the inorganic forms of the element.
It should be emphasised again that LOQ in these examples is just a guideline as to the real-world detection capability of the technique for this method. However, it does offer a very good approximation as to whether the technique is suitable based on the factor improvement number compared to the J-values for each elemental impurity.
Table 1 shows that axial-ICP-OES offers definite possibilities for monitoring Class 2A elements in oral drugs, but apart from cadmium, which has an improvement factor of 500, the technique might struggle with the Class1 elements, lead, arsenic and mercury because the improvement factors are all less than 6. These numbers could be further improved, by using a much higher sample weight or lower dilution volumes in the sample preparation procedure without compromising the method. Alternatively, As could be determined by hydride generation, while Hg could be quantified using the cold vapour technique.
Table 1. USP Chapter <232> J-values for Class 1 and 2A elements compared to limits of quanitation for axially-viewed ICP-OES
However, it can be seen in Table 2 that ICP-MS shows significant improvement factors for all the Class 1 and 2A impurities. Even for the four heavy metals, there appears to be ample improvement to monitor them with good accuracy and precision. The added benefit of using ICP-MS is that it would also be suitable for the other methods of pharmaceutical delivery, such as parenteral or inhalation, where the PDE levels are typically one or two orders of magnitude lower. Additionally, if arsenic or mercury levels were found to be higher than the PDE levels, it would be relatively straight-forward to couple HPLC with ICP-MS to monitor the speciated forms of these elements if required.
Table 2. USP J-values for Class 1 and 2A elements compared to limits of quanitation for ICP-MS
The current PDE limits described in USP Chapter <232>, and ICH Q3D Step 4 guidelines, together with validation protocols described in Chapter <233>, presents unique challenges in order to show suitability of the analytical procedure being used. From a practical standpoint, there is no question that to meet the PDE limits in all pharmaceutical delivery methods, particularly for parenteral and inhalation drugs where the PDE’s are significantly lower, ICP-MS is probably the most appropriate technique. However for oral delivery products, especially liquid medications or those that can be easily brought into solution with a suitable aqueous or organic solvent, axial-ICP-OES could offer a more cost-effective approach. In addition, ICP-OES can use larger sample weights and lower dilutions, which will improve its detection capability. However, ICP-MS has shown it has the detection limits and throughput capability, and coupled with HPLC for speciated forms of As and Hg, it appears to be the optimum technique of choice for carrying out the measurement of elemental impurities in a wide and diverse range of pharmaceutical materials.
1. Elemental Impurities in Pharmaceuticals: Updates: USP Website: http://www.usp.org/chemical-medicines/elemental-impurities-updates
2. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html.
3. Measuring Elemental Impurities in Pharmaceuticals: A Practical Guide, R.J. Thomas, CRC Press, Boca Raton, FL, ISBN:-13: 978-1-138-19796-1, February, 2018
Robert J. Thomas has worked in the field of trace element analysis for over 40 years, including 24 years for an ICP-MS manufacturer and 15 years as a principal of his own consulting company. He has served on the American Chemical society (ACS) Reagent Chemical Committee for the past 17 years as leader of the elemental impurities task force where he has worked closely with the United States Pharmacopeia (USP) to align heavy metal testing procedures in reagent chemicals with those of pharmaceutical materials. He has authored almost 100 publications on trace element analysis and written three textbooks on ICP-MS and related topics, including this new book, which focuses on the new global directives on elemental impurities in pharmaceutical materials. He is currently editor and frequent contributor to the Atomic Perspectives Column in Spectroscopy Magazine. He has an advanced degree in Analytical Chemistry from the University of Wales in the UK and is a Fellow of the Royal Society of Chemistry (FRSC, and a Chartered Chemist (CChem).
Lab Asia 33.4 - August 2026




.jpg)









-(1).jpg)

.jpg)