Microscopy & microtechniques
Published over 10 years ago. See the latest and most current information on Microscopy & microtechniques.
Electron microscopy delineates a detailed topology of cells. However, temporal information is largely missing in electron micrographs. To visualize membrane dynamics, we have developed a novel technique, ‘flash-and-freeze’, that induces particular cellular activity with a flash of light and captures membrane trafficking events at defined time points after the induction by rapid freezing of cells. A time series of snap shots allows reconstruction of a ‘flip-book’ of membrane dynamics. Using this technique, we have characterised membrane trafficking events at neuronal synapses that occur on a millisecond time scale. The flash-and-freeze approach therefore adds a missing piece of information to electron micrographs – time.
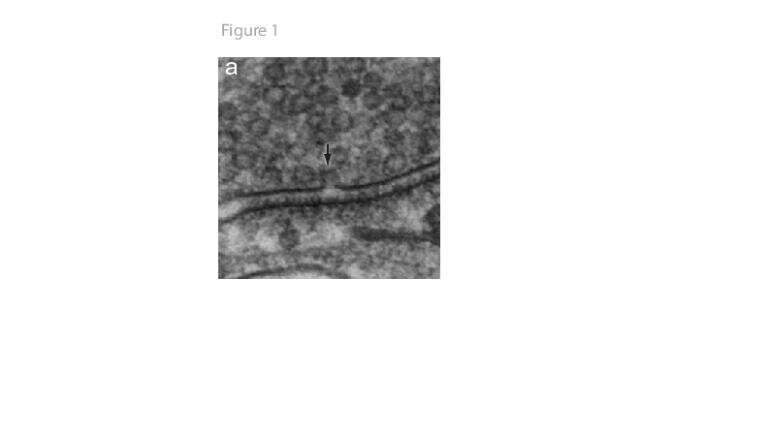
A picture is worth hundreds of words. However, a snap-shot can be deceiving. In an electron micrograph of a synaptic terminal (Figure 1), a membrane invagination is observed among the cluster of synaptic vesicles. Because the size of the invagination is roughly equal to the size of a synaptic vesicle, it is tempting to conclude that this structure represents fusion of a synaptic vesicle. However, it is equally possible that the membrane pit represents an endocytic intermediate. Therefore, it is imperative to know what happened before and what happened afterwards in order to deduce what is going on in a picture.
Traditionally, electron microscopy only captures a static image of cells – cells are fixed prior to imaging. Fixation comes in two flavours: chemical cross-linking and cryo-immobilization. First, lipids and proteins can be cross-linked using chemicals such as paraformaldehyde and glutaraldehyde. These chemicals diffuse into cells, fixing lipids and proteins in place by cross-linking them. The chemical fixation is generally a slow process, and it induces morphological changes as a result of the chemical reaction between the cellular constituents and the fixatives used, as well as osmotic alterations. Alternatively, cells can be immobilized by rapid freezing. Freezing is a physical process, lacking the drawbacks induced by the chemical fixation. In either case, the cells are “fixed” while they are performing their natural functions. Thus, occasionally, interesting structures might be captured, but the nature of these structures cannot be fully determined from these images.
One potential solution to this problem is to induce a particular cellular function and fix cells at defined time points from the induction [1]. The chronological compilation of images enables the reconstruction of a flip-book of the cellular event. How can a particular cellular function be induced? In the recent years, light appears to be the solution. Optogenetics is a technique that uses heterologously-expressed light-sensitive proteins to control particular cellular functions [2]. For example, neuronal activity can be induced by activation of light-sensitive cation channels, channelrhodopsin [3]. Lipid composition of plasma membrane can be modified by light-induced recruitment of phosphatidylinositol phosphatase to the plasma membrane [4]. Likewise, many cellular functions can be induced by a flash of light. These techniques can create the time zero in cells of interest.
Following the induction of cellular activity, cells must be fixed at the precise time points. To achieve the precise control, we employed a high-pressure freezing technique. High-pressure freezing can immobilize cells instantaneously while minimising the artifact caused by ice crystal formation [5]. However, in the commercial high-pressure freezer, cells or tissues are embedded within a closed compartment, and thus light cannot reach the specimens. Therefore, we modified the existing sample holder and created the light path (Figure 2) [6,7]. This modification allowed us to control the time intervals between light stimulation and freezing at milliseconds time scale and visualize membrane dynamics in electron micrographs.
Using this approach, now termed ‘flash-and-freeze’, the Jorgensen laboratory and the Watanabe laboratory are studying membrane trafficking events at neuronal synapses. For neuronal communication, synaptic vesicles are consumed rapidly at synaptic terminals. These vesicles must be recycled locally at synapses because a limited number of synaptic vesicles is available at each terminal and synaptic terminals are located far away from the cell body where the components of synaptic vesicles are synthesised. Based on the morphological studies in the ‘70s and ‘80s, two models for synaptic vesicle recycling were put forward. One model suggests that synaptic vesicles are regenerated slowly via clathrin-mediated endocytosis [8]. Another model suggests that synaptic vesicles are recycled rapidly via reversal of the fusion process [9]. Despite the extensive research over the last 40 years, there is no resolution to this controversy.
To test how synaptic vesicles are recycled, we stimulated mouse hippocampal neurons, expressing channelrhodopsin, and froze them at defined time points ranging from 15 ms to 20 s (Figure 3) [6,10,11]. At 15 ms, synaptic vesicles are captured in process of fusing with the plasma membrane. Exocyotsis of synaptic vesicles only occurred in the region, called active zone, which is juxtaposed to the post-synaptic receptive field. At 30 ms, those fusing vesicles are flattened out into the membrane. By 50 ms, no membrane invagination was observed in the active zone. Instead, shallow invagination was observed in the region just outside of the active zone. These invaginations progressively turned into vesicles of about 80 nm in diameter and were fully internalised by 100 ms. The internalized vesicles represent the endocytic intermediates of synaptic vesicles as they are delivered to endosomes, from which synaptic vesicles are regenerated via clathrin scaffolds. These results suggest that synaptic vesicle recycling is a two-step process: rapid internalization of excess membrane followed by slower regeneration of synaptic vesicles from endosomes [12]. The flash-and-freeze approach is thus very powerful for studying membrane dynamics at millisecond temporal resolution.
Recently, this approach became more accessible. A high-pressure freezer, EM ICE, from Leica Microsystems**(Figure 4), offers synchronised high-pressure freezing and light stimulation to reveal these intricate changes in fine structure and cellular dynamics.
With the development in optogenetic tools, essentially any cellular functions can be manipulated by light in the near future. The flash-and-freeze approach allows visualization of membrane trafficking events underlying these cellular functions at a millisecond temporal resolution and a nanometer spatial resolution. What comes before? What comes after? Obtaining the temporal information in your electron micrograph is within the reach.
Figure 1. An electron micrograph of a synapse from mouse hippocampal neuronal cultures. An arrow indicates membrane invagination captured in the active zone.
Figure 2. Schematic diagrams showing the modified specimen holder of the high-pressure freezer, EM PACT2, with LED turned off (a) and on (b). Specimens are embedded within the metal cup, indicated by yellow. The specimen holder is modified to house an LED inside the holder. A sapphire anvil is placed at the end of the screw to allow light transmission. Wayne Davis in the Jorgensen Laboratory designed the sample holder and wrote a program to interface the light stimulation device with the high-pressure freezer.
Figure 3. Electron micrographs of synapses at defined time points after stimulation. (a-d) Exocytosis of synaptic vesicles are observed during the first 30 ms of the stimulus. (e,f) Endocytosis initiates at 50 ms post stimulation and completed by 100 ms after stimulation (g-j). (k,l)The internalized vesicles move away from the plasma membrane (300 ms post stimulus).
Figure 4. (a) Erik Jorgensen and Shigeki Watanabe and (b) with the R&D team involved in the instrument development.
1. Heuser, J. E. & Reese, T. S. Structural changes after transmitter release at the frog neuromuscular junction. J. Cell Biol. 88, 564–580 (1981).
2. Fenno, L., Yizhar, O. & Deisseroth, K. The development and application of optogenetics. Annu. Rev. Neurosci. 34, 389–412 (2011).
3. Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G. & Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 (2005).
4. Idevall-Hagren, O., Dickson, E. J., Hille, B., Toomre, D. K. & De Camilli, P. Optogenetic control of phosphoinositide metabolism. Proc. Natl. Acad. Sci. U. S. A. 109, E2316–2323 (2012).
5. Moor, H. in Cryotechiniques in Biological Electron Microscopy 175–191 (Springer-Verlag, 1987).
6. Watanabe, S. et al. Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. eLife 2, e00723 (2013).
7. Watanabe, S., Davis, M. W. & Jorgensen, E. M. in Nanoscale Imaging of Synapses (eds. Nägerl, U. V. & Triller, A.) 43–57 (Springer New York, 2014).
8. Heuser, J. E. & Reese, T. S. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J. Cell Biol. 57, 315–344 (1973).
9. Ceccarelli, B., Hurlbut, W. P. & Mauro, A. Depletion of vesicles from frog neuromuscular junctions by prolonged tetanic stimulation. J. Cell Biol. 54, 30–38 (1972).
10. Watanabe, S. et al. Ultrafast endocytosis at mouse hippocampal synapses. Nature 504, 242–247 (2013).
11. Watanabe, S. et al. Clathrin regenerates synaptic vesicles from endosomes. Nature 515, 228–233 (2014).
12. Watanabe, S. Slow or fast? A tale of synaptic vesicle recycling. Science 350, 46–47 (2015).
For further details on EM ICE please contact [email protected]
Lab Asia 33.4 - August 2026



.jpg)









-(1).jpg)



.jpg)










