Laboratory products
Per- and polyfluoroalkyl substances (PFAS) pose a unique analytical challenge. Their extreme chemical stability, ubiquity at ultra‑trace levels, and prevalence across water, food, and environmental matrices require methods that are both highly sensitive and intrinsically robust. Liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS), operated in multiple‑reaction monitoring (MRM) mode, has become the cornerstone of targeted PFAS analysis. In parallel, simplified extraction approaches, often described as blend‑and‑inject, are redefining laboratory throughput by shifting reliance from extensive clean‑up toward instrumental selectivity and matrix tolerance. This article translates core LC–MS/MS principles into practical guidance, illustrating how contamination control, delay‑column strategies, and streamlined sample preparation can deliver reliable PFAS data across waters, foods, soils, biosolids, and food‑contact materials.
PFAS comprise a diverse family of fluorinated surfactants valued for their resistance to heat, oil, and water. These same properties underpin their environmental persistence and bioaccumulation, driving regulatory scrutiny and the need for defensible, low‑level measurements. From an analytical perspective, PFAS test the limits of routine workflows. They are often present at parts‑per‑trillion (ppt) concentrations, readily adsorb to common laboratory materials, and are ubiquitous in solvents, tubing, seals, and even laboratory air.
As a result, PFAS analysis is not solely about sensitivity; it is about background management, method discipline, and robustness under real‑world conditions. LC–MS/MS has emerged as the preferred platform because it combines chromatographic separation with molecular specificity and exceptional sensitivity. When paired with thoughtful contamination control practices and efficient extraction schemes, LC–MS/MS enables high confidence quantitation without sacrificing throughput.
This article focuses on translating principles into practice. Rather than emphasising a particular instrument brand or commercial workflow, it highlights general device architectures, ionisation concepts, and sample handling strategies that have proven effective across routine laboratories handling water, food, and environmental solids.
Where PFAS appear, and why it matters
Modern PFAS monitoring spans drinking and surface waters, wastewater effluents, biosolids, soils, foods (such as eggs, fish, edible oils, and teas), and food‑contact materials including paper wraps and sachets. Each matrix presents distinct analytical complications. Water samples demand ultra‑low detection limits; foods introduce fats, proteins, and surfactants; soils and sludges contain humic material; and consumer packaging can release unexpected extractables.
Crucially, these challenges influence every stage of the workflow - from the choice of labware and solvents to chromatography and detection. Failure to account for PFAS adsorption or background contamination can undermine even the most sensitive mass spectrometer.
Core device architecture
For targeted PFAS quantitation, triple‑quadrupole LC–MS/MS remains the workhorse. Operated with electrospray ionisation (ESI) in negative mode, PFAS typically form stable [M–H]⁻ precursor ions. Tandem MS allows selection of a specific precursor ion in the first quadrupole, fragmentation in a collision cell, and monitoring of characteristic product ions in the third quadrupole.
This multiple reaction monitoring (MRM) approach provides two decisive advantages: selectivity against matrix interferences and quantitative precision at low ppt and sub‑ppb levels. By monitoring both a quantifier and qualifier transition, laboratories gain orthogonal confirmation beyond retention time alone.
Breadth of application
The same LC–MS/MS principles extend beyond a single matrix. Panels of PFAS can be measured at ppt levels in water, at sub‑ppb levels in complex foods, and across a broad carbon‑chain range in soils and biosolids. Importantly, the platform also supports other polar contaminants such as pharmaceuticals or oestrogens, reinforcing the value of a unified analytical approach for modern environmental and food testing laboratories.
From molecules to measurements
A typical LC–MS/MS workflow integrates sample introduction via liquid chromatography, atmospheric‑pressure ionisation, ion transport into vacuum, and mass filtering across three quadrupoles. Software extracts chromatograms corresponding to specific precursor‑to‑product transitions and applies calibration models for quantitation.
MRM is especially powerful for PFAS because many compounds share similar backbones and chemical properties. Gas‑phase fragmentation and transition‑specific monitoring effectively substitute for extensive sample clean‑up, filtering away chemical noise and enabling confident reporting even in complex extracts.
Background and contamination control
Because PFAS are present in instrument components and laboratory consumables, contamination control is not optional, it is foundational. Best practices include the routine use of polyethylene or polypropylene labware, avoidance of Polytetrafluoroethylene (PTFE) where feasible, and the use of nitrile gloves not only for safety but also as a contamination barrier.
A particularly effective tactic is the installation of a short reverse‑phase delay column upstream of the analytical column between the pump and injector. This device retains PFAS originating from the system itself, separating these background contributions from genuine sample analytes and preventing incorrect peak assignment at low concentration levels.
Ionisation and matrix tolerance
Instrument interfaces designed with heated desolvation, orthogonal sampling, and laminar‑flow ion guides improve tolerance to complex matrices. These designs minimise the deposition of non‑volatile material, reduce the frequency of cleaning, and preserve signal‑to‑noise across long analytical sequences—an essential prerequisite for simplified extraction workflows.
Traditional approaches
Broad multiresidue workflows have traditionally relied on QuEChERS‑type extractions, combining solvent partitioning, salts, and dispersive solid‑phase cleanup. While effective, these approaches can be time‑consuming, particularly when freezing or multiple clean‑up steps are required for fatty matrices. There is also an element of increased risk of potential contamination which could impact the results generated.
For aqueous samples, solid‑phase extraction (SPE) has similarly served as a cornerstone technique, offering concentration and cleanup in a single workflow. Yet its strengths come with practical limitations: SPE is intrinsically labour‑intensive, requires multiple conditioning and elution steps, and can become difficult when dealing with waters containing high particulate loads, such as those sourced from rivers, lakes, or surface runoff. Filtration, cartridge clogging, and extended processing times can significantly slow throughput and introduce variability. These constraints highlight why more streamlined, matrix‑tolerant approaches, such as direct injections or simplified solvent extractions, are increasingly valuable in modern PFAS surveillance, where both sensitivity and operational efficiency are essential.
The blend‑and‑inject concept
Blend‑and‑inject represents a strategic shift. Samples are homogenised, extracted with acetonitrile or methanol, centrifuged, and directly injected often within 30–45 minutes. Rather than relying on extensive cleanup to protect the instrument, the approach leverages chromatographic separation and MRM selectivity to maintain data quality.
This minimalist preparation has proven effective for avocados, eggs, fish, fish oil, teas, food‑contact papers, soils, and biosolids. When paired with matrix‑tolerant LC–MS/MS hardware and appropriate contamination control, blend‑and‑inject can dramatically increase throughput without compromising quantitation.
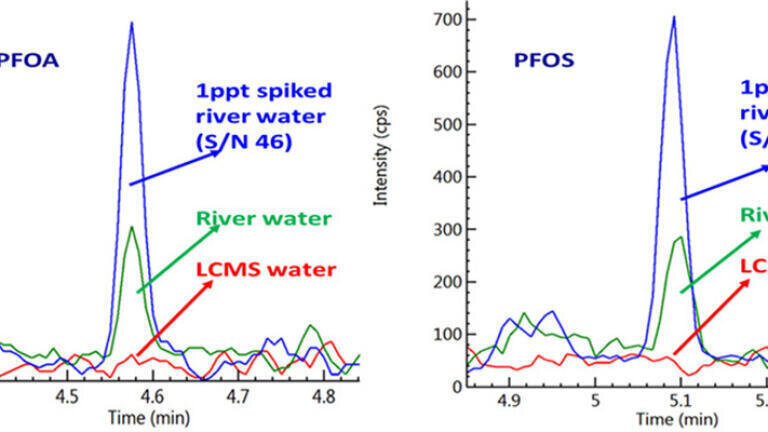
Waters at the ppt Level
Figure 1: Analysis of PFOA and PFOS in water at 1ppt level.
Targeted PFAS panels have been quantified in natural and drinking waters at low‑ppt concentrations. Delay columns efficiently separate background system PFAS from sample peaks, enabling confident identification and quantitation of compounds such as PFOA and PFOS at approximately 1 ppt.
Foods: Eggs, Fish, Oils, and Teas
Figure 2: Performance of PFAS compounds in Fish Oil (Omega-3) at 0.25ppb.
Sub‑ppb PFAS concentrations have been measured in eggs using rapid acetonitrile extraction and direct injection. Even lipid‑rich matrices such as fish oil yield strong signal‑to‑noise ratios when supported by robust ion optics and optimised gradients. Tea samples illustrate rapid comparative screening, with PFAS detected in some products but absent in others under identical conditions.
Food‑contact materials
Figure 3: Food Contact Material Analysis across various packaging types.
Simple solvent extractions of paper‑based food packaging have revealed PFAS such as PFHpA. Here, the confirmatory power of dual‑transition MRM is essential, reducing false positives in matrices rich in paper additives and extractables.
Biosolids and soils
Figure 4: 12 PFAS compounds were confirmed in the biosolid sample (quantifier/qualifier ions shown.
Methanolic extraction combined with LC–MS/MS enables the detection of PFAS across a wide chain‑length range in sludges and soils. Short‑chain acids, long‑chain carboxylates, sulfonates, and fluorotelomer acids can all be confirmed through ion‑ratio agreement, supporting site assessments without protracted sample preparation.
Identity and quantitative integrity
Robust PFAS data rely on multiple layers of confirmation. Quantifier and qualifier ion ratios provide a powerful identity check, particularly in the presence of ubiquitous background contamination. Matrix‑matched calibration or isotope dilution further improves accuracy and linearity across wide concentration ranges, from sub‑ppt in waters to low‑ppb in foods.
Desolvation efficiency, orthogonal sampling geometries, and laminar‑flow ion guides collectively enhance robustness. These design features sustain sensitivity during long sequences of complex samples, enabling thousands of injections with minimal maintenance. Such resilience is what allows streamlined prep strategies to scale beyond proof‑of‑concept into routine operation.
Compared with immunoassays, LC–MS/MS offers compound‑level specificity and confirmatory capability. Relative to GC‑based approaches, it avoids derivatisation for most polar PFAS. While high‑resolution MS excels in non‑target discovery, triple‑quadrupole MRM remains unmatched for routine, targeted quantitation and delivers the highest sensitivity levels available.
Successful PFAS workflows align preparation, chromatography, mass spectrometry, and QA/QC. Key elements include background‑aware lab practices, delay‑column usage, matrix‑appropriate extraction, and disciplined use of confirmatory ion ratios. When these principles are applied together, laboratories can achieve rapid turnaround without sacrificing confidence.
All PFAS data presented in this article were acquired using the PerkinElmer QSight® 500 LC/MS/MS System, a triple‑quadrupole platform engineered for high‑throughput, contamination‑resistant analysis of complex matrices. The system’s direct‑injection capability and StayClean™ technology support the streamlined, blend‑and‑inject workflows described throughout this article. The QSight 500 was recognised as Innovation of the Year at the Labmate Awards for Excellence 2025, cited for its ability to deliver robust PFAS quantitation with minimal sample preparation and reduced downtime.
PFAS analysis exemplifies the intersection of chemistry and engineering. Ultra‑trace targets, complex matrices, and pervasive background contamination demand methods that are selective, robust, and pragmatic. Triple‑quadrupole LC–MS/MS, combined with contamination aware practices and blend‑and‑inject sample preparation, meets this challenge effectively.
As regulatory attention intensifies and PFAS target lists expand, these foundational principles will remain central. By trusting instrument selectivity, managing background proactively, and right‑sizing sample preparation to the analytical question, laboratories can deliver what stakeholders ultimately need, fast, confirmatory, and defensible answers about what is truly present in the sample.
Lab Asia 33.4 - August 2026



.jpg)










-(1).jpg)


.jpg)





.jpg)




.jpg)
