Research news
A recent review has synthesised evidence to demonstrate that immune ageing – marked by thymic involution, chronic low-grade inflammation, and immunosenescence (aging-related changes to the immune system) – acts as a significant accelerator of type 2 diabetes (T2DM) in older populations. The authors described how age-related immune decline interacts with cellular stress pathways to disrupt the so-called ‘ominous octet’ of eight interconnected organ dysfunctions that perpetuate hyperglycaemia, and consequently identifying potential targets for therapeutic intervention.
“Immune ageing is not merely a bystander but a catalytic force in the progression of T2DM,” the authors stated. They argued that integrating immunometabolic stress mechanisms into this established framework offers a route to preserve pancreatic β-cell function and immune resilience.
The review highlighted several mechanisms that link immune ageing to T2DM. Chronic low-grade inflammation, or ‘inflammaging’, results from the senescence-associated secretory phenotype, which releases pro-inflammatory cytokines such as interleukin-6 and tumour necrosis factor alpha. This process impairs insulin signalling, worsens insulin resistance and induces β-cell death through oxidative stress and endoplasmic reticulum dysfunction. Ageing also shifts macrophage activity from anti-inflammatory to pro-inflammatory phenotypes, further destabilising metabolic balance.
The authors detailed how compensatory hyperinsulinaemia, initially a protective response, becomes pathological. Elevated insulin levels activate stress kinases such as c-Jun N-terminal kinase and nuclear factor kappa B, which disrupt insulin receptor signalling and reduce glucose uptake. This fuels a self-reinforcing cycle in which inflammation and metabolic dysfunction accelerate β-cell exhaustion.
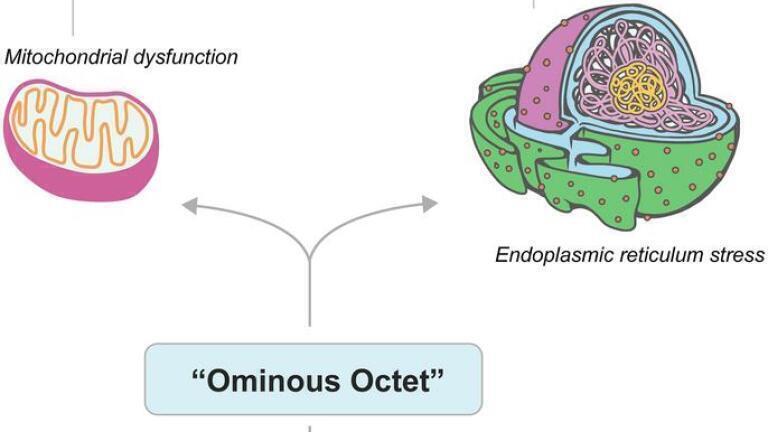
Organelle dysfunction emerged as a unifying pathway in disease progression. Mitochondrial impairment reduces adenosine triphosphate synthesis, increases reactive oxygen species, and disrupts calcium signalling, hampering insulin secretion and promoting excessive glucose production by the liver.
Endoplasmic reticulum stress leads to protein misfolding, activation of the unfolded protein response and inhibition of insulin receptor trafficking and glucose transporter type 4 translocation. Persistent stress triggers β-cell apoptosis via pro-inflammatory signalling pathways.
These immune and cellular stress mechanisms were shown to exacerbate all elements of the so-called ‘ominous octet’, including:
The authors proposed multi-targeted strategies to interrupt this immune-metabolic cycle. Suggested interventions include senolytic agents such as dasatinib and quercetin to remove senescent cells, specialised pro-resolving mediators to resolve inflammation, and glucagon-like peptide-1 receptor agonists to promote anti-inflammatory macrophage activity.
Approaches to protect organelle function – including mitophagy enhancement, unfolded protein response regulation and stabilisation of mitochondrial-endoplasmic reticulum contact sites – were also recommended. Biomarkers such as C-reactive protein, interleukin-6, and serine-phosphorylated insulin receptor substrate-1 were identified as possible tools to personalise treatment.
The review concluded that future research should account for differences in age and ethnicity and explore emerging fields such as gut microbiome-immune interactions, circadian rhythm disruption, and α- to β-cell transdifferentiation. The authors emphasised that a mechanistic understanding of immune ageing could enable the development of therapies that preserve metabolic control in ageing populations.
For further reading please visit: 10.14218/ERHM.2025.00018
Lab Asia 33.4 - August 2026



.jpg)









-(1).jpg)