Research news
Cold Spring Harbor Laboratory researchers have reported that a bespoke antisense oligonucleotide can disrupt alternative splicing of Aurora kinase A, which has collapsed a feedback loop that links SRSF1, Aurora kinase A and MYC in pancreatic ductal adenocarcinoma cells and has reduced cell viability while it has triggered apoptosis
Pancreatic ductal adenocarcinoma, often abbreviated to pancreatic ductal adenocarcinoma (PDAC), is widely regarded as the deadliest major form of pancreatic cancer and it accounts for most diagnoses of the disease. Many experimental and approved targeted approaches in pancreatic cancer have sought to inhibit Kirsten rat sarcoma viral oncogene homologue, commonly known as KRAS, because mutations in this oncogene drive tumour initiation and maintenance in a large proportion of PDAC cases.
However, clinicians have faced persistent obstacles, not least because tumours can resist single-agent pressure through redundant signalling routes and adaptive stress responses. Researchers have therefore continued to pursue combination strategies and alternative vulnerabilities that might help to overwhelm these defences.
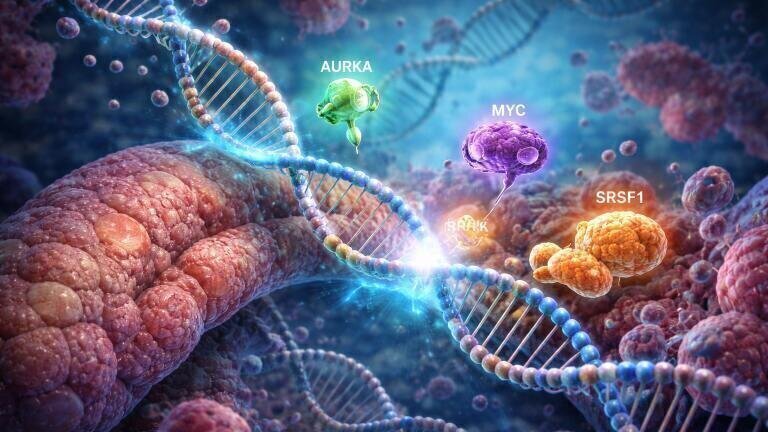
A team at Cold Spring Harbor Laboratory (CSHL), Long Island, New York, USA, led by former graduate student Alexander Kral in the laboratory of Professor Adrian Krainer, has revisited data from a 2023 study that linked the splicing factor SRSF1 to PDAC tumour development. That earlier work suggested that elevated SRSF1 could ‘jumpstart’ tumorigenesis. The recent analysis has indicated that SRSF1 acts as part of a three-component regulatory circuit that also involves Aurora kinase A (AURKA) and another major oncogene, MYC, which together appear to promote aggressive disease progression.
“Our theory was that some of the changes caused by increased levels of SRSF1 were playing a role in the accelerated tumour growth we were seeing,” Kral said.
“We homed in on a molecule we thought could be an important driver of this called Aurora kinase A (AURKA). We found it’s part of a complex regulatory circuit that includes not only AURKA and SRSF1, but another key oncogene called MYC,” he added.
In mechanistic terms, the researchers reported that SRSF1 influenced AURKA via alternative splicing, a process that allows cells to generate distinct messenger RNA isoforms from a single gene by selective inclusion or exclusion of exons. In cancer, shifts in alternative splicing can rewire protein output without any change to the underlying DNA sequence. Here, the group described how SRSF1-dependent splicing changes increased AURKA production. The resulting AURKA then stabilised the MYC protein, which is often difficult to inhibit directly because it lacks the sort of binding pocket that small-molecule drugs usually require. MYC, in turn, raised SRSF1 levels, which restarted the cycle and created a self-reinforcing loop that could help tumour cells to sustain high oncogenic drive.
“Bits and pieces of this circuit were known previously, but we didn’t have the full picture until now,” Krainer said.
“Once we figured out alternative splicing of AURKA was involved, we could start looking into ways to disrupt it,” he said.
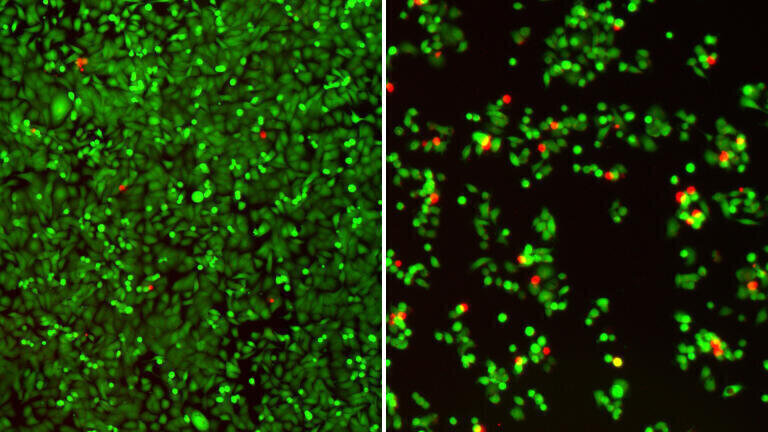
To test that idea, the team designed a bespoke antisense oligonucleotide, a short synthetic strand of nucleic acid that can bind a specific RNA sequence and redirect splicing choices. Antisense oligonucleotides, often shortened to antisense oligonucleotides (ASOs), are a longstanding speciality of the Krainer laboratory, which previously contributed to the development of nusinersen – marketed as Spinraza – an antisense oligonucleotide that the United States Food and Drug Administration (FDA) approved as a treatment for spinal muscular atrophy. In the PDAC system, the researchers aimed to use their antisense oligonucleotide to obstruct the particular AURKA splicing pattern that supported the circuit.
According to the team, the effect went beyond partial interference. By shifting AURKA splicing, the antisense oligonucleotide collapsed the wider SRSF1–AURKA–MYC loop, which reduced tumour cell viability and activated apoptosis, the tightly regulated form of programmed cell death that healthy tissues use to remove damaged or dangerous cells.
“It’s like killing three birds with one stone. SRSF1, AURKA, and MYC are all oncogenes contributing to PDAC progression. Just by targeting AURKA splicing with our ASO, we see the loss of these other two molecules as well,” Krainer said.
The researchers have emphasised that translation to the clinic remains distant. Antisense therapies must meet demanding requirements for stability, delivery to tumour tissue, on-target activity and acceptable toxicity, and pancreatic tumours present particular delivery challenges because dense stromal tissue can limit drug penetration. Even so, the work has offered a clear rationale to pursue splicing modulation as a route to hit multiple oncogenic drivers at once, which could complement efforts to target KRAS or to combine KRAS inhibition with additional mechanisms.
The Krainer laboratory has continued to refine the antisense oligonucleotide with a view to improve potency and to define the safest route to administration. The underlying message is familiar to anyone who follows translational science: basic mechanistic work often lays the first stones of therapies that appear years later.
For further information please visit: 10.1016/j.molcel.2025.12.004
Lab Asia 33.4 - August 2026



.jpg)









-(1).jpg)