Clinical
Researchers at The Wistar Institute have engineered a small-molecule drug conjugate that has directed an Aurora kinase A inhibitor into tumours via heat shock protein 90 targeting, increased intratumoural exposure, reduced detectable toxicity in preclinical models and improved tumour control when paired with WEE1 inhibition
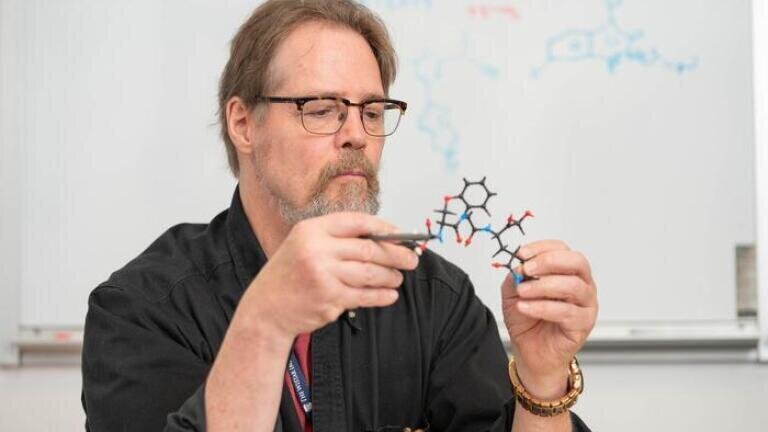
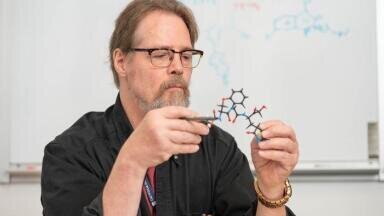
Scientists at The Wistar Institute, Philadelphia, Pennsylvania, USA, have reported a preclinical strategy to improve a well-established class of anti-cancer agents by coupling tumour targeting with cell-cycle inhibition in a single chimeric compound. The approach has combined an Aurora kinase A inhibitor with a heat shock protein 90-targeting moiety to create a small-molecule drug conjugate designed to increase drug exposure inside tumours while limiting exposure in healthy tissue.
Aurora kinase A inhibitors have attracted sustained interest because Aurora kinase A regulates mitotic progression and supports malignant proliferation, yet dose escalation has often proved difficult in practice because off-target exposure can produce toxicity in non-malignant cells. In this study, investigators wanted to examine if they could improve the therapeutic index by re-routing the inhibitor towards tumour tissue.
“An Aurora kinase A inhibitor is viewed as a lethal synthetic molecule in cancer therapy, but the problem is you can’t dose it high enough, because then it starts to spill over and [affect] normal cells, causing toxicity,” said Dr. Joseph Salvino, co-author of the study and professor in Wistar’s molecular and cellular oncogenesis programme.
“By using this … targeted approach [of a molecule already in clinical use], we can direct it to cancer cells, increasing its exposure in the tumour itself,” he said.
The conjugate has joined two functional components in a modular design. The first component has inhibited Aurora kinase A, a kinase central to orderly chromosome segregation and cell division. The second component has bound heat shock protein 90, a molecular chaperone that tumours frequently express at high levels to maintain proteome stability under metabolic and microenvironmental stress. By exploiting this differential expression pattern, the team aimed to enrich the therapeutic payload in malignant tissue.
In proof-of-concept experiments, the chimeric molecule has demonstrated engagement of both biological targets. In cancer cell models with cells derived from head and neck malignancies, lung cancer and melanoma, the treatment has been shown to block proliferation and replication and has led to cell death. These findings have supported the central mechanistic premise that simultaneous target recognition can preserve anti-proliferative activity while changing tissue distribution.
The investigators then moved to preclinical in vivo models to assess pharmacokinetics and tumour exposure. They reported that intratumoural concentrations of the conjugate reached levels up to ten times higher than those achieved with the parent Aurora kinase A inhibitor alone.
They also found prolonged tumour retention, with measurable activity at 24 hours post-administration, whereas the unconjugated comparator had become undetectable by that time. The compound was well tolerated in the tested models and the study reported no significant toxicity signal under experimental conditions.
When researchers paired the conjugate with a WEE1 inhibitor, tumour-growth control improved further, consistent with the knowledge base in the wider literature on coordinated disruption of cell-cycle checkpoints. That result has suggested potential for combination regimens in settings where monotherapy cannot sustain durable response.
“‘When drugs fail in the clinic, half the time it’s because of poor exposures in the tumour due to pharmacokinetic problems,” Salvino said, in reference to limitations in absorption, distribution and effective tissue penetration.
“‘Our approach will take an existing compound and improve its pharmacokinetic properties, enhancing its exposures in the tumour,” he added.
The authors have argued that the platform could extend beyond the initial tumour types and support adaptation to other payloads where efficacy is constrained by systemic toxicity or inadequate target-site exposure. That proposition fits an established translational theme in oncology drug development – to optimise delivery and tumour pharmacology to reinvigorate the use of already active mechanisms.
Next steps have included plans to test analogous conjugates across additional cancer contexts and to develop an oral formulation for the current molecule. If successful, oral delivery could expand clinical flexibility, support chronic scheduling where appropriate and simplify combination use.
For further reading please visit: 10.1158/1535-7163.MCT-25-0857
Lab Asia 33.4 - August 2026












-(1).jpg)







-(1).jpg)
